-
Torahiko Yamaguchi, Yohsuke Yamamoto, Daisuke Kinoshita, Kin-ya Akiba, ...
Session ID: O-01
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
The precursor allenes
1a (R = Me) and
1b (R = Ph) were synthesized. The dimethylation of
1 afforded the corresponding sulfonium salt
2, which is hypervalent hexacoordinate carbon species. Combination of methyl iodide with a silver salt afforded single crystals of
2, that was suitable for an X-ray analysis. The X-ray structure of
2b indicated that the distance between the oxygen atom of the methoxy group and the central carbon atom was shorter than that of the phenoxy group. Furthermore, synthesis of the triplet carbene by two-electron oxidation of
1a was attempted.
View full abstract
-
Makoto Yamashita, Yasutomo Segawa, Yuta Suzuki, Yu Takeuchi, Kyoko Noz ...
Session ID: O-02
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
The reaction of boryllithium and borylmagnesium, which are boryl anion equivalent, were examined. Boryllithium could react with alkyl halides to form the corresponding alkylborane and borylmagnesium did not react with these alkyl halides. In the reaction with benzaldehyde having C=O double bond, boryllithium was converted to borylbenzylalcohol and borylmagnesium was derived to benzoylborane. On the other hand, boryllithium could react with azines possessing C=N double bond to afford the borylated azines in good yields.
View full abstract
-
Hiroshi Yamaoka, Kyoko Okada, Kimio Isa, Yoshio Takai, Nico M.M. Nibbe ...
Session ID: O-03
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
Unimolecular dissociation for a variety of deuterium labeled N, N'-diacylimidazolidine-2-thiones has been reported during metastable time window using a double reversed geometry (BE/BE) four sector tandem mass spectrometer (JEOL JMS-700T). Competitive channels such as remote double-single/triple hydrogen transfer processes have been opened originated from two benzyl hydrogen atoms of the acyl group during metastable window under both the high energy process (10 keV) and high resolution mass selection as well as high mass resolution product ion scanning (R= 1000/1000, 10% valey conditions in both precursor and product ions).
View full abstract
-
Yasushi Ohga, Aiichiro Takami, Toru Takahashi, Tsutomu Asano
Session ID: O-04
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
Pressure effects of photochromism of napthopyran having dimethyl, diphenyl, adamantylidene, and fluorenylidene substituents at 2-position were studied in ethanol, methyl acetate, methylcyclohexane, 2-methylpentane-2,4-diol (MPD), glycerol triacetate (GTA), and 2,4-dicyclohexyl-2-methylpentane (DCMP). In highly viscous solvents (MPD, GTA, and DCMP), viscosity induced retardation, i.e. dynamic solvent effect, was observed at higher pressures. The effect of bulkiness of substituents at 2-position on the dynamics of solvent reorganization will be discussed on the basis of pressure and viscosity effects.
View full abstract
-
M.N. Kumara, Teshu Nakahara, Masaaki Mishima
Session ID: O-05
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
Second-order rate constants k
N for the reactions of diarylmethyl cations with alcohols, amines, and oximes in acetonitrile solution were measured by the laser flash photolysis. Using the Mayr's equation log k
N = s(E + N), the N and s were determined for these nucleophiles. The N values decrease in the order of
n-BuOH >
n-PrOH > EtOH > MeOH >
i-PrOH > H
2O >
t-BuOH. The N values of alcohols in acetonitrile are considerably larger than the corresponding solvent nucleophilicity for second-order rate constants. It is found that there exists a linear relationship between the N values and gas-phase basicities excluding bulky nucleophiles. In addition, nucleophilicities of oximes are higher than the expected ones based on their basicities, suggesting the existence of α-effect for these nucleophiles.
View full abstract
-
Takaaki Sonoda, Magdalena Pasikowska, Masaaki Mishima, Taizo Ono, Haru ...
Session ID: O-06
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
In the course of our computational and experimental studies about the fluorine effect on the structure and properties of various kinds of strong and superstrong acids1), we report here our recent results of B3LYP/6-311+G(d,p) level computational study and FT-ICR (Ion Cyclotron Resonance) MS experimental study about the gas phase acidity of some polyfluorinated noncyclic and caged carbon acids and oxygen acids.The significant conclusion drawn is that 1-H-perfluoroadamantane(1-H-F-adamantane) and 1-HO-perfluoroadamantane(1-HO-F-adamantane) are endowed with an intrinsic gas phase acidity close to 320 and 315 kcal mol
-1, respectively, similar to that of conventional strong acids such as nitric acid and methane sulphonic acid, respectively, and exceeded over that of HC(CF
3)
3. It is remarkable that the acidities of C
10F
21OH and F-triamantane derivative C
18F
23OH are as strong as super acid (CF
3SO
2)
2NH.
View full abstract
-
Ryoichi Akaba
Session ID: O-07
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
In order to compare the relative stability (the ease of formation) of peroxy radical cations, which are not isomeric to each other, we utilize the isodesmic reactions in which only difference in both sides is to which olefin radical cation molecular oxygen adds. By calculating energy with B3LYP/6-31G(d) level of theory of the equilibrium structures of various peroxy radical cations with use of a peroxy radical cation as a standard, we can quantitatively compare the relative stability of various peroxy radical cations.
Extended data set of various peroxy radical cations containing phenyl, alkyl, and adamantyl groups are presented, and the role of the substituent effects on the stability of these reactive species will be discussed.
View full abstract
-
Kenzi Hori, Hirotaka Sadatomi, Hidetoshi Yamamoto
Session ID: O-08
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
We have been investigating this concept, called In Silico screaning of synthesis routes, by combining theoretical calculations and chemoinformatics. A mitsunobu reaction, one of the synthesis routes for 2,6-dimethylchroman-4-one 1, which the KOSP program created, was examined using the DFT calculations at the B3LYP/6-31+G level of theory. We found a transition state structure which leads to the key intermediate of Mitsunobu reaction from a phenolate derivative ion. Another TS for the side product was also found although the energy difference between the two TSs is higher than that expected from the experimental result. These mechanisms are consistent with results from synthesis experiments.
View full abstract
-
Daisuke Kaneno, Shuji Tomoda
Session ID: O-09
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
L-Selectride undergoes preferential equatorial attack at cyclohexanones. However, it has long been known that the facial diastereoselection of the Danishefsky pyranones (2-methoxy-2,3,5,6-tetrahydro-4-pyranones) with L-Selectride is the opposite.
Based on the exterior frontier orbital extension model previously proposed, we have found that the ground state conformational and the LUMO properties should be the origin of the observed 'unusual' facial selection. The EFOE data suggested: (1) the axial face of the 2,3,5,6-tetrahydro-4-pyranone ring system is much less sterically hindered than the corresponding face of cyclohexanone owing to the deformation of the six-membered ring upon replacement of the carbon by an oxygen at the 4-position and (2) the EFOE data of minor conformers can also account for the experimental results.
View full abstract
-
Satoshi Kitaoka, Kaoru Nobuoka, Yasushi Ohga, Yuichi Ishikawa
Session ID: O-10
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
In general, rearrangement of solvent molecules is more rapid than movement of a reactant solute. However, this relation can be inverted when viscosity of the used solvent drastically increase by applying high pressure. Here we firstly report this phenomenon, known as the dynamic solvent effect, could be observable even in imidazolium ionic liquids. Pressure effect (0.1 M-600 Mpa) on a photoisomerization reaction in an ionic liquid was monitored from the viewpoint of the molecular structure of the salt medium. We would discuss the relation between the dynamic solvent effect and the salt design (side chain structure of the imidazolium cation and anion size).
View full abstract
-
Satoshi Usui, Mutsuo Okamura
Session ID: O-11
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
Theoretical studies on the ks-kD competition solvolysis of 2-phenyl ethyl system was carried out by using PCM solvation energy calculation . The reaction potential energies for the kD process obtained in the gas phase indicated the phenonium ion intermediate structure 5 kcal/mol more stable than that of the reactant. The PCM solvation energy calculation in water exhibited that the phenonium ion intermediate is 13.5 kcal/mol less stable than the reactant, and is 1.5 kcal/mol more stable than the transition state. Thus, the PCM solvation energy calculation successfully afforded the reaction potential energy expected for the concept of the normal organic reaction.
The theoretical ks/kD ratio obtained in the gas phase exhibited the kD preference while in water ks is preferred over kD that is in the same line in the solvolysis.
View full abstract
-
Hiroshi Ikeda, Hayato Namai, Nobuyui Kato, Kazuhiko Mizuno
Session ID: O-12
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
Substituent effects on the energies (Eob) of electronic transitions of geminally diphenyl-substituted trimethylenemethane (TMM) radical cations
2•+, the excited state of the corresponding biradical
2••*, and those of structurally related 1,1-diarylethyl cations
3+ and radical
3• were determined experimentally by using electronic transition spectroscopy. In addition, transition energies of these reactive intermediates were determined by using density functional theory (DFT) and time-dependent (TD)-DFT calculations.
View full abstract
-
Kazuko Nakazono, Toshikazu Takata
Session ID: O-13
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
It has been difficult to neutralize the ammonium of rotaxane having sec-ammonium salt and crown ether. This time we succeeded in the
N-methylation of the sec-ammonium of rotaxane. The obtained tert-ammonium-type rotaxane was neutralized to novel tert-amine-type rotaxane by base. We will talk about the mechanism and the property of tert-ammonium and amine type rotaxane.
View full abstract
-
keiji Hirose, Yamato Nakamura, Hirokazu Takano, Yoshito Tobe
Session ID: O-14
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
Two series of rotaxanes, which consist of axles with amide group and rings with phenol moiety, have been synthesized by aminolysis of prerotaxanes. The kinetics of the deslipping reaction of these rotaxnes were studied. The deslipping of the rotaxane having nitro group attached to the phenol moiety proceeds at 100 degrees. The reactions of rotaxanes, which have hydrogen or bromine substituent at the corresponding position, did not occur even at 120 degrees. These results can be interpreted by the difference in intercomponent hydrogen bonds. We also revealed steric effect on the deslipping reactions. The deslipping rates of diastereomeric rotaxanes, which have chiral center both the ring and the axle, are obviously different. In nonpolar solvents, the difference of deslipping rates
was originated from the different activation enthalpy. On the other hand, in the polar solvents, difference of deslipping rates was originated from the different activation entropy.
View full abstract
-
Koji Sawai, Tsukasa Nakahodo, Hisashi Fujihara
Session ID: O-15
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
Metal nanoclusters and their nanocomposites containing nanoscale organic and inorganic materials have drawn a lot of attention because they exhibit interesting optical, electronic, and catalytic properties. Control of the surface properties and reactivities on metal nanoclusters is an important aspect of developing nanomaterial applications. The size, shape, and surface properties of metal nanoclusters are crucially controlled by the nature of protective ligands. Previously we reported that optically active bisphosphine, 2,2'-bis(diphenylphosphino)-1,1'-binaphthyl [BINAP]-stabilized palladium nanoclusters catalyzed asymmetric hydrosilylation. This paper presents the synthesis of chiral phosphine-stabilized metal nanoclusters and their metal nanocluster-catalyzed asymmetric Suzuki-Miyaura cross-coupling reactions.
View full abstract
-
Yuko Iiduka, Koji Nakajima, Takatsugu Wakahara, Tsukasa Nakahodo, Taka ...
Session ID: O-16
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
Endohedral metallofullerenes have attracted special attention as new spherical molecules with unique properties unexpected from empty fullerenes. Among thes, scandium metallofullerenes are of special interest because of the high variety in fullerene size as well as their relatively high yields. Discussion will be focused on the electronic and chemical property of scandium metallofullerenes.
View full abstract
-
junpei YUASA, Shunichi FUKUZUMI
Session ID: O-17
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
Perchloric acid promotes hydride reductions of p-quinone and o-quinone derivatives by an NADH analogue. The hydride reductions proceed via electron transfer from the NADH analogue to the quinone derivatives. The mechanism of hydride reductions will be disccused.
View full abstract
-
Satoshi Ogawa, Masahiro Yoshida, Sayuri Inoue, Kazuaki Shimada
Session ID: O-18
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
We synthesized benzannulated spirotelluranes bearing chalcogen ligands by the reactions of dichalcogenastannoles with 0.5 equiv. of tellurium tetrachloride. The crystal structure of these molecules were characterized by X-ray crystallography. The behavior and thermal reactions of the spirotelluranes in solution were observed by
1H NMR spectroscopy and product analyses.
View full abstract
-
Morifumi Fujita, Sakuro Okuno, Hee Jin Lee, Takashi Sugimura, Tadashi ...
Session ID: O-19
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
Reactions of oxy-functionalized alkenylboronates with hypervalent iodine(III) reagent gave alkenyliodonium salts with complete inversion of configuration. The substitution reaction proceeds via a 1,3-dioxan-2-yl cation intermediate generated by the participation of internal carbonyl oxygen.
The participation of internal oxy groups was also observed for the reaction of alkenylsilanes. The reaction of alkenylsilane changed the reaction mode from substitution to oxygenation under the control of the participation. The triethylsilyl group remained in the oxygenated product, while the boronate group was eliminated. Participation of the acyloxy group during the reaction of alkenylsilyanes with iodosylbenzene provides a-silyl ketone and 2-silyltetrahydrofuran without elimination of the silyl group. The reaction proceeds via 1,3-dioxan-2-yl cation intermediate similar to the reaction of alkenylboronate
View full abstract
-
Akihiko Ishii, Masayuki Ohishi, Norio Nakata
Session ID: O-20
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
(1-Adamantyl)-t-butyldithiirane 1,2-dioxide and di-t-butyldithiirane 1,2-dioxide were synthesized by oxidation of the corresponding dithiirane 1-oxides with dimethyldioxirane. The thermolysis of dithiirane 1,2-dioxides produced the sulfines and SO in the main path. It was verified that the generated SO reacted with thioketones to give the dithiirane 1-oxides. The mechanism of the thermolysis was also considered with DFT calculations. The reaction of dithiirane 1,2-dioxides with a platinum(0) complex gave the disulfenato Pt(II) complexes. Oxidation of the related sulfenato-thiolato Pt(II) complex was investigated.
View full abstract
-
Shunsuke Furukawa, Junji Kobayashi, Takayuki Kawashima
Session ID: O-21
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
The Friedel-Crafts reaction is well known as a method for introducing the substituent directly into the aromatic ring. However, Sila-Friedel-Crafts reaction involving a silyl cation as a reactive intermediate has not been reported as a versatile synthetic method. On the other hand, siloles are useful compounds, which are applied to optical materials. Herein, we describe the development of intramolecular Sila-Friedel-Crafts reaction and its application to the synthesis of benzosiloles.
View full abstract
-
Taro Tanabe, Yoshiyuki Mizuhata, Norihiro Tokitoh
Session ID: O-22
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
Syntheses, structures, and properties of silanedichalcogenolato platinum complexes,
2a-
c, which were prepared by utilizing stable silanedichalcogenols
1a-
c as key building blocks, will be described in this presentation. When silane(oxylato)chalcogenolato-platinums
2b-
c were treated with HCl in the presence of H
2O,
2b-
c underwent the β-Mes elimination to give the dihydroxysilanechalcogenolato-platinums
3b-
c respectively. Five-coordinate silicates are most likely suggested as plausible intermediates, which should be formed by the nucleophilic attack of a chloride ion toward the silicon atom of the initially generated hydroxo complexes. This is the first example of the direct observation of β-aryl elimination from metal siloxides.
View full abstract
-
Masayasu Igarashi, Masaaki Ichinohe, Akira Sekiguchi
Session ID: O-23
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
The cyclotrisilene (
1), (
tBu
2MeSi)
2SiSi
2(Si
tBu
3)
2, was synthesized by the reaction of 2 equiv of (
tBu
2MeSi)
2SiLi
2with
tBu
3SiBr
2SiBr
2Si
tBu
3 in THF. The cyclotrisilene
1 was reacted with Ph
3C
+Ar
4B
- in dried and degassed toluene at room temperature for 8 h to produce cyclotrisilenylium ion (
2+).
The adamantyl-substituted disilacyclopropene (
3) was synthesized by the reaction of bis(tri-tert-butylsilyl)dilithiosilane with 1-adamantanecarbonyl chloride. The disilacyclopropene
3 was reacted with Ph
3C
+Ar
4B
- in dried and degassed toluene at room temperature for one minute to produce cyclotrisilenylium ion (
4+).
View full abstract
-
Tetsuzi Katori, Hiroshi Yamataka
Session ID: P-01
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
For the Schmidt reaction, it is expected that not only rearrangement but also fragmentation reaction takes place depending on substituent as in the Beckmann rearrangement. In this study, we analyzed pathways of the Schmidt reaction of substituted benzyl methyl ketones by theoretical calculations to clarify how a change of a reaction pathway occurred. It was found that as substituent became electron donating, reaction pathway changed from rearrangement to fragmentation as in the Beckman rearrangement. In addition, in iminodiazonium ion which was an intermediate of the Schmidt reaction a structure that was not obtained for the oxonium ion, an intermediate of the Beckman rearrangement, was found. We were able to find new pathways of the rearrangement and fragmentation reactions.
View full abstract
-
Ryo Akimoto, Hiroshi Yamataka
Session ID: P-02
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
Schmidt rearrangement reaction of aldehydes and ketones in strongly acidic media containing hydrazoic acid is a versatile approach to nitriles and amides.
The reaction gives a fragment product aside from a rearrangement product like amide, when a displacement atom group exists stably in reaction solution. So substituted 3-phenyl-2-butanone have the stable atom group were synthesized and allowed to react with trimethylsilyl azide in trifluoroacetic acid. After the reaction, the existence of the two products of rearrangement and fragmentation was determined. In addition, to clarify a reaction mechanism of the Schmidt reaction, a correlation between a product ratio and reaction is examined.
View full abstract
-
hiroto hasegawa, hiroshi yamataka
Session ID: P-03
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
The Beckmann rearrangement takes place when ketoxime is treated with acid, but a fragmentation reaction occurs when the migrating atom group is stable as the cation. Theoretical calculations with 3-phenyl-2-propanoneoxime have suggested that both rearrangement and fragmentation reactions proceed through a common transition state. To prove the results of the theoretical calculations by experiment, 3-aryl-2-propanoneoxime 2-naphthalenesulfonates and 3-aryl-2-butanoneoxime 2-naphthalenesulfonates were synthesized and the reactivity and the ratio of the rearrangement and the fragmentation product were determined.
View full abstract
-
Atsuki YANAGISAKO, Kaoru NOBUOKA, Satoshi KITAOKA, Yuichi ISHIKAWA
Session ID: P-04
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
The Diels-Alder reactions in imidazolium ionic liquids(ILs) showed that the hydrogen bond between dienophile and the imidazolium cation via the C2-H affected the
endo/exo stereoselectivity of the product. In particular, the counterion of the imidazolium cation is known to have an influence on the hydrogen bond.
In this study, we carried out Diels-Alder reaction in the ionic liquids possessiong cyano groups.
View full abstract
-
Takao Okazaki, Kenneth K. Laali
Session ID: P-05
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
Arenediazonium tetrafluoroborate salts undergo metathesis on immobilization in 1-butyl-3-methylimidazolium bis(trifluoromethanesulfonato)amide [BMIM][Tf
2N]. The "noncoordinating", "nonnucleophilic" [Tf
2N] anion acts as an ambident nucleophile towards the aryl cations, formed via thermal dediazoniation, to give predominantly the oxy anion quenching products [ArO-SO(CF
3)=NTf], with minimal formation of ArN(Tf)
2, irrespective of the nature of the substituent(s) on the ArN
2+. Strong preference for the formation of oxygen trapping products did not change under photolytic conditions, where dediazoniation occurs at room temperature. Minimal amount of the Schiemann product ArF is also formed in both thermal and photolytic dediazoniation, depending on the substituent(s). Progress of dediazoniation in the IL (both thermal and photolytic), and the evolution of the products were directly monitored by
1H and
19F NMR.
View full abstract
-
Yoh-Hei Yabuno, Manabu ABE
Session ID: P-06
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
The Paterno-Buchi (PB) reaction of furyl methanol derivative with benzophenone was performed in this study to understand the effect of hydrogen-bonded interaction on the regio- and stereoselectivity in the oxetane formation reactions. We found that in polar solvents, the trans-configured oxetane was selectively formed, but the temperature effect on the regioselectivity was very small.
View full abstract
-
Yoshitomo Takeuchi, Yui Araki, Mutsuo Okamura
Session ID: P-07
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
Generally, the coenzyme NAD
+-NADH does stereoselective redox reactions of substrate by a hydride transfer at C
4 of NAD ring. As in application, we have tried to synthesize the coenzyme NAD
+ analogues with CF
3 group at C
4, and examined the face-seletivity of CF
3 transfer reaction on trifluoromethylated analogues (CF
3-NADH).
We synthesized CF
3-NADH by a CF
3 addition of CF
3 anion to NAD
+ analogue. Against the purpose, attack of anion to C
2 of NAD+ analogues took first priority, 3-carbamoyl-1,2,4-trimethyl-2-CF
3-1.2-dihydroquinoline (2-adduct) have been provided. In this trifluoromethylation, the face-selectivity was observed for the orientation of carbamoyl at C
3. Two rotational isomers of 2-adduct were also observed around the C
3-carbonyl bond and slowly exchaneged. The ratio of isomers changes with various solvents.
View full abstract
-
Mari Iizuka, Hitomi Oosaki, Masato Yoshida, Takaaki Sonoda
Session ID: P-08
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
We have been investigating the methods for the synthesis of various fluorine compounds using the reaction of perfluoroalkyl iodide with α-methylstyrene. In this conference, we would like to report the synthetic methods for α-methylstyrenes possessing various fluoroalkyl groups on the α-methyl carbon by photochemical reaction of perfluoroalkyl iodide with α-methylstyrene in the presence of titanium oxide. The formation of this thermodynamically less stable olefin is discussed on the basis of MO calculation. The titanium oxide-catalyzed trifluoromethylation with trifluoromethyl iodide was however unsuccessful. Thus, as an alternative method, photochemical oxygenative trifluoromethylation of α-methylstyrene with trifluoromethyl iodide in the presence of ditin was developed to give the olefin via the corresponding alcohol. Further, the reactions of these olefins for their synthetic utility were investigated.
View full abstract
-
Kazumasa Kajiyama, Daisuke Itou, Maiko Mineshiro, Takeshi Miyamoto
Session ID: P-09
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
The effective radical additions of the bis(8-oxy-1-naphthyl) hydrophosphorane to several unsaturated compounds could be achieved by activation with 1,1'-azobis(cyclohexanecarbonitrile) (ACHCN). The reactions were regioselective and the major isomers of the resulting alkenes were Z-alkenes. In the absence of ACHCN the addition reactions of the hydrophosphorane to the unsaturated compounds also proceeded.
View full abstract
-
Sakuro Okuno, Hee Jin Lee, Morifumi Fujita, Takashi Sugimura, Tadashi ...
Session ID: P-10
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
We recently found that the reaction of 4-acyloxybut-1-enyl(triethyl)silanes with iodosylbenzene gave 3-acyloxy-2-(triethylsilyl)tetrahydrofuran. The oxygenation of the acyloxy substrate can be explained by intermediate formation of 1,3-dioxan-2-yl cation, which is trapped by water leading to the tetrahydrofuran. The tetrahydrofuran formation was also observed for the reaction of simple acyloxybutenes. Optically active hypervent iodine(III) reagents were newly prepared and employed for the tetrahydrofuran formation reactions. The enantioselectivity was related to structure of acyloxyalkene substrates.
View full abstract
-
Md. Mizanur Rahman Badal, Masaaki Mishima
Session ID: P-11
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
Acidities of oximes are particularly interesting, for their conjugate anions are referred to as a-nucleophiles. To clarify intrinsic nature of the anions, the substituent effects on the gas phase acidity of acetophenone oxime have been studied. The gas phase acidities were determined based on proton transfer equilibria using a Fourier transform ion cyclotron resonance (FT-ICR) spectrometer. The gas phase acidities of acetophenone oximes are related linearly with the corresponding acidities of phenols with a slope of 0.80 including the
p-NO
2 group, indicating a significant pi-delocalization of the negative charge formed at the oxygen atom of the oximes into the aromatic pi-system. This is consistent with the geometrical features of the oximate ions and the charge distribution obtained by the theoretical calculations at B3LYP/6-31+G* level of theory.
View full abstract
-
shuhei itoh, masaaki mishima
Session ID: P-12
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
In this study, dimethoxy boron cation basicities (BCB) were determined for pyridines by measuring the ligand exchange equilibrium using an FT-ICR mass spectrometer. Plot of relative BCBs of pyridines against the corresponding gas phase basicities (GB) toward protons shows a good linear relationship for 3-, 4-substituted pyridines with a slope of 0.81 for pyridines indicating that the substituent effect on the stability of the boron cation complexes is similar to that for corresponding protonated species. This suggests that the bonding interactions between dimethoxy boron cation and these organic compounds have a highly covalent character. In addition, large negative deviations from the line based on 3-, 4-substituted pyridines are observed for 2-substituted pyridines. These deviations are due to steric inhibition of π-interaction between π-orbital of pyridines and empty p-orbital of boron cation, being consistent with computational results at B3LYP/6-311+G(d,p) level.
View full abstract
-
Tomotaka Miyazaki, Ikuma Hayakawa, Daisuke Akie, Katsuyuki Hirai, Tosh ...
Session ID: P-13
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
Thiol molecules are adsorbed on the gold substrate and form self-assembled monolayers (SAM). We previously synthesized trithiol (Molecular Tripod)
1 containing CH
2SH groups at the three bridgehead positions of adamantane framework, and demonstrated that this molecule forms a SAM with regular molecular arrangement with three-point absorption on gold substrate. In this work, we synthesized the trithiol connected with ferrocene (
2) and triptycene (
3), which will allow observation of reactions and conformational changes of the molecules fixed on the substrate.
View full abstract
-
Tadashi Akasaki, Yangsoo Lee, Koichi Komatsu, Toshikazu Kitagawa
Session ID: P-14
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
A dichloroethyl adduct (CH
3CCl
2-C
60-Cl) and a chloromethyl adduct (ClCH
2-C
60-Cl) of C
60 were obtained by the reaction of AlCl
3 and C
60 in dichloromethane. In this research, the dichloroethyl adduct was converted to fullerenol (CH
3CCl
2-C
60-OH) by hydrolysis. The cation, generated by dissolving the fullerenol in triflic acid, could be observed by NMR.
The structure and stability of this cation was compared with other previously reported alkylated C
60 cations.
View full abstract
-
Toshikazu Takata, Atsuyoshi Tarutani, Hisahiro Sasabe
Session ID: P-15
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
A [2]rotaxane was synthesized from a secondary ammonium salt axle having [60]fullerene and a crown ether wheel possessing a porphyrin moiety via the end-capping of the corresponding pseudo[2]rotaxane formed initially. When a mixture of a [60]fullerene-containing crown ether wheel and a porphyrin-containing crwon ether wheel was used, [3]rotaxane having mixed wheel components was selectively obtained with use of a bis(secondary annmonium salt) axle. The selective formation of the hetero [3]rotaxane was attributed to the attractive interaction between the fullerene and porphyrin moieties.
View full abstract
-
Satoru Sato, Yutaka Maeda, Koji Inada, Tadashi Hasegawa, Michio Yamada ...
Session ID: P-16
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
Endohedral metallofullerenes have attracted broad attention because of their novel properties due to the intramolecular interaction between the metal atom and the fullerene cage. New electronic properties, such as the low oxidation and reduction potentials, induced by the interaction would allow new application of fullerenes. Reversible addition reaction is useful method for separation of fullerenes and protection of their reactive sites. Recently, we have reported the reversible and regioselective addition reaction of La@C
82 with cyclopentadiene (Cp). This retro-reaction proceeds much faster than that of C
60Cp. In this context, it is important to retard the retro-reaction of La@C
82Cp. It has been reported that an adduct of C
60 and pentamethylcyclopentadiene (Cp*) is less prone to undergo the retro-reaction than C
60Cp. Herein, we report the reversible addition reaction of La@C
82 with Cp* and the stability of its adduct.
View full abstract
-
Takeshi Nakamura, Akinobu Takegami, Manabu ABE
Session ID: P-17
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
Singlet biradicals are key intermediates in processes involving a bond-cleavage and bond-formation. Normally the species are short-lived and it is quite difficult to characterize the reactivity by experiments. In this study, we found a long-live 2-silapropane-1,3-diyl that has a lifetime to react intermolecularlly with PTAD.
View full abstract
-
Shigeyoshi Inoue, Masaaki Ichinohe, Akira Sekiguchi
Session ID: P-18
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
Tetrasilyldisilene (
tBu
2MeSi)
2Si=Si(SiMe
tBu
2)
2 (
1) undergoes chemical oxidation with Ph
3C
+ TPFPB
- and Et
3Si
+ TPFPB
- (TPFPB
- = tetrakis(pentafluorophenyl)borate). When a mixture of
1 and 1.1 equivalent of Ph
3C
+ TPFPB
- in toluene was stirred at room temperature over 30 min, the disilene cation-radical
2 was isolated as its borate salt in 85% yeild as red brown, air- and moisture-sensitive crystals.
Furthermore,
1 react with 1.1 equivalent of Et
3Si
+ TPFPB
- to give cyclic trisilaallyl cation
3 via elimination of
tBu
2MeSi and Me group,
3 was isolated as its brate salt in 78% yield as red crystals.
View full abstract
-
Teruyo Ikeda, Hiroshi Ikeda, Masafumi Yamada, Yasutake Takahashi, Seig ...
Session ID: P-19
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
A photoinduced electron-transfer reaction of 3,4-bis(α-styryl)furan in which the initially formed radical cation undergoes cyclization to pruduce tetramethylene (TME)-type radical cation. This followed by back electron transfer to give TME type biradical. These new TME type intermediates were observed using transient absorption spectroscopy on laser flash photolysis. Moreover, we gained insight into the multiplicity of observed biradical with quantum chemical calculation.
View full abstract
-
Hiroaki Kotani, Toshiya Ono, Kei Ohkubo, Shunichi Fukuzumi
Session ID: P-20
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
Photoirradiation of a deaerated mixed solution (2.0 mL) of phthalic acid buffer (pH 4.5; 50 mM) and MeCN [1 : 1 (v/v)] containing Acr
+-Mes and NADH results in formation of the Acr
•-Mes. Upon addition of Pt-PVP to a deaerated solution of Acr
•-Mes, efficient ET from Acr
•-Mes to Pt-PVP occurs via proton-coupled ET because the log values of the ET rate constants are plotted as a function of pH. In addition, the inverse isotope effect was observed in the case of deuterated buffer solution. This indicates that the rate-determining step is formation of Pt-H bonding.
View full abstract
-
Ryosuke Iwata, Kei Ohkubo, Shunichi Fukuzumi
Session ID: P-21
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
Addition of 9,10-dimethylanthracene to an acetonitrile solution of [Ru(bpy)
3]
3+ (bpy = 2,2'-bipyridine) as an oxidant exhibited a broad absorption band at NIR region (> 2000 nm). Such a broad NIR absorption is diagnostic of the pai-dimer radical cation formed form pai-complexation between neutral anthracene and its radical cation. On the other hand, the pseudo-first order rate constants (
k1) of one-electron oxidation of 9-bromoanthracene with [Fe(bpy)
3]
3+ was monitored by stopped-flow spectrophotometer. The
k1 value increases with an increase in anthracene concentration to exhibit a second-order dependence because of formation of pai-dimer radical cation. In summary, anthracene can act as a self-catalyst in the electron-transfer oxidation of anthracene by formation of anthracene dimer radical cation.
View full abstract
-
Mohammad Gulam Rabbani, Toru Takahashi, Yasushi Ohga
Session ID: P-22
Published: 2007
Released on J-STAGE: October 06, 2008
CONFERENCE PROCEEDINGS
FREE ACCESS
Photochromism of zinc dithizonate was studied in order to explore the dynamic solvent effects on the photochromic isomerization. A solution of zinc dithizonate undergoes color changes from pink to colorless upon irradiation with visible light and the pink color returns after cut off irradiation. The return process was followed spectrophotomeriacally at high pressures in low viscosity solvent, methyl acetate, and high viscosity solvent, glycerol triacatete. The rate constants decrease slightly with increasing pressures in methyl acetate. On the other hand, in glycerol triacetate, viscosity-induced retardations were observed at higher pressures. Since these retardations of the reactions were observed only at high viscosities, they were taken to indicate a failure of the transition state theory caused by slow solvent thermal fluctuations. Pressure, viscosity, and temperature effects on this isomerization will be discussed on the basis of two dimensional reaction coordinate model.
View full abstract

 Download PDF (325K)
Download PDF (325K) Download PDF (98K)
Download PDF (98K) Download PDF (278K)
Download PDF (278K) Download PDF (48K)
Download PDF (48K) Download PDF (37K)
Download PDF (37K) Download PDF (721K)
Download PDF (721K) Download PDF (35K)
Download PDF (35K) Download PDF (25K)
Download PDF (25K) Download PDF (195K)
Download PDF (195K) Download PDF (136K)
Download PDF (136K)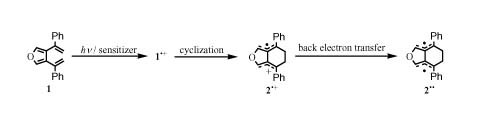 Download PDF (37K)
Download PDF (37K)